Medical Treatment for ALS
|
|
|
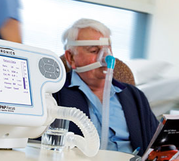
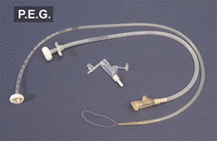
Currently, the treatment of ALS relies on sensitive communication and education of the diagnosis and symptoms, involvement of the patient and their family, a positive care plan, and ongoing evaluation and intervention with a multidisciplinary team (Radunović, Mitsumoto, & Leigh, 2007). Drug treatment using Riluzole has been effective in improving the survival rate about three to six more months. However, providing respiratory therapy such as home ventilation and use of a bimodal positive airway pressure (BiPap) device as soon as vital capacity declines to 50% has been the key in prolonging survival rate and improving quality of life. Medications for spasticity and musculoskeletal discomfort may be prescribed. Since maintenance of adequate nutrition is a problem for many patients with ALS, introducing a PEG tube early will result in better nutrition and hydration. According to Langmore et al., (2006), there is tentative evidence for a nutritional and survival advantage in patients with PEG tubes.
As ALS progresses, a decline in muscle strength is often seen. Therefore, collaborating with occupational and physical therapists in early management of muscle weakness will be important to preserve patient’s mobility and ambulation. Lightweight plastic orthoses may help to support or facilitate patient’s movements. Selection of a wheel chair seating system must adapt in response to the patient’s disease progression. Additionally, it is important to be aware that seating/positioning arrangements of the patient will have an impact on decision-making on the control site and placement of Augmentative Communication systems. Exercising can serve to maintain joint range of motion and prevent uncomfortable contractures. Additionally, various types of adaptive equipment can address the problem of decreased mobility and improve ability to perform activities of daily living. This may include electric mobility devices with various means of control, or environmental control units with switches to allow access to television, lights, and telephones (Yorkston, 2010).
References:
Langmore, S. E., Kasarskis, E. J., Manca, M. L., & Olney, R. K. (2006). Enteral tube feeding for amyotrophic lateral sclerosis/motor neuron disease. The Cochrane database of systematic reviews, (4), CD004030. doi:10.1002/14651858.CD004030.pub2
Radunović, A., Mitsumoto, H., & Leigh, P. N. (2007). Clinical care of patients with amyotrophic lateral sclerosis. The Lancet Neurology, 6(10), 913–925. doi:10.1016/S1474-4422(07)70244-2
Yorkston, K. M., Beukelman, D. R., Strand, E. A., & Hakel, S. (2010). Management of motor speech disorders in children and adults. Austin: Pro-Ed.
As ALS progresses, a decline in muscle strength is often seen. Therefore, collaborating with occupational and physical therapists in early management of muscle weakness will be important to preserve patient’s mobility and ambulation. Lightweight plastic orthoses may help to support or facilitate patient’s movements. Selection of a wheel chair seating system must adapt in response to the patient’s disease progression. Additionally, it is important to be aware that seating/positioning arrangements of the patient will have an impact on decision-making on the control site and placement of Augmentative Communication systems. Exercising can serve to maintain joint range of motion and prevent uncomfortable contractures. Additionally, various types of adaptive equipment can address the problem of decreased mobility and improve ability to perform activities of daily living. This may include electric mobility devices with various means of control, or environmental control units with switches to allow access to television, lights, and telephones (Yorkston, 2010).
References:
Langmore, S. E., Kasarskis, E. J., Manca, M. L., & Olney, R. K. (2006). Enteral tube feeding for amyotrophic lateral sclerosis/motor neuron disease. The Cochrane database of systematic reviews, (4), CD004030. doi:10.1002/14651858.CD004030.pub2
Radunović, A., Mitsumoto, H., & Leigh, P. N. (2007). Clinical care of patients with amyotrophic lateral sclerosis. The Lancet Neurology, 6(10), 913–925. doi:10.1016/S1474-4422(07)70244-2
Yorkston, K. M., Beukelman, D. R., Strand, E. A., & Hakel, S. (2010). Management of motor speech disorders in children and adults. Austin: Pro-Ed.
